TL;DR
This 2014 PNAS paper by S. Lin et al (Lin et al., PNAS, 2014) that compares transcription of tissues between species has a flawed experimental design, where species is almost perfectly confounded with machine / lane on which the sequencing was done. Y. Golad and O. Mizrahi-Man have published a manuscript describing the confounding and the results of removing it. This was possible because the original authors supplied the information about which publically available files were used in the original analysis. The data from this experiment is probably only suitable as an example of what not to do in high-throughput biology experimental design, and that there may be similarities in human and mouse transcriptional programs.
We discussed these papers in the Systems Biology and Omics Integration University of Kentucky Journal Club on June 1, 2015. I lead that discussion.
Mouse / Human Transcriptomic Differences
In 2014, two papers were published by members of the ENCODE project purporting that tissue gene expression clustered more by species than by tissue. In the first (ENCODE Consortium, Nature, 2014, doi:10.1038/nature13992 2014), a large number of experiments were combined and compared. Figure 2a shows a PCA plot of expression in across 10 tissues in human and mouse.

ENCODE paper figuer 2
Interestingly enough, if you collapse PC1 (definitely species), then the tissues start to look quite similar. Which does not seem that unexpected, there are species specific differences, but the tissues are doing something similar in each species.
This was not the end of the story, however. A subsequent publication (S Lin et al., PNAS, 2014, doi: 10.1073/pnas.1413624111) went further in doing fresh sequencing of 13 tissues in both species, still showing bigger differences between species than between tissues.
Is Everything As it Seems?
On April 28, Y. Gilad sent out this tweet:
We reanalyzed the data from http://t.co/Fv7z9WwLJ4 and found the following: pic.twitter.com/37eVs8Kln9
The left figure shows the original data, clustering by species, and on the right, reprocessed data, clustering by tissue. Now, I have to admit when he posted this I was kind of ticked off. Where was the blog-post or manuscript showing what exactly was done? If you look at the comments to Yoav on that tweet, it seems others were wondering the same thing. Thankfully, on May 19, the manuscript hit F1000Research (Gilad Y and Mizrahi-Man O. A reanalysis of mouse ENCODE comparative gene expression data [v1; ref status: approved with reservations 1, http://f1000r.es/5ez] F1000Research 2015, 4:121 (doi: 10.12688/f1000research.6536.1)).
Batch Effects
Yoav asked for the list of data files that were used in the PNAS paper, and then examined the read id line to extract the experimental design. This experimental design is captured in Figure 1:

Y Golad Fig 1
Do you notice a problem with this design?
. . . . .
Hopefully you noticed that species (one of the main effects to investigate) is not randomly or even semi-randomly distributed across sequencers and / or lanes, but is almost perfectly confounded with sequencer / lane. It doesn’t matter if one technician handled all the samples, this is potentially a large batch effect / confounding variable.
And Yoav shows that ignoring the batch effect produces data much like that reported in the Lin et al PNAS pub, while removing the batch effect using ComBat results in the tissues clustering together.
Is the Data Still Useful?
I presented these papers and the discussion around them at our weekly Systems Biology and Omics Integration (SBOI) Journal Club on June 1, 2015. There were a couple of concerns with the overall study:
- Is ENCODE data still useful?
- some people seemed to be concerned that this brought the general ENCODE project into question. I think by the end we agreed that in general it is probably ok to be using ENCODE data, but this particular study was questionable.
- What does the data tell us?
- based on the correction that Yoav did and reanalysis, is there anything actually telling in the data? Unfortunately, because species is so confounded with machine, I don’t think there is much of a conclusion to draw about species differences vs tissue similarity based on this data. There was some disagreement on this point.
- Full experimental designs should be published, and requested by reviewers
- Really, why a reviewer on the paper did not request more information about the experimental design is beyond me, especially given the claims of the manuscript. Why it is not the norm to provide this kind of experimental design information in a manuscript is also a good question.
Finally, I think the best use of the 2014 PNAS pub and this dataset is an example of how not to design a biological experiment.
Addendum
As I looked at the comment section of the Gilad & Man article yesterday (June 1, 2015), I noticed that there were direct replies from S. Lin in a couple of places. In particular is a comment that Lin et al did a second set of sequencing with a new design, and reanalysed the data. Links are provided to two figures, a table of the new design and a new 3D PCA plot:
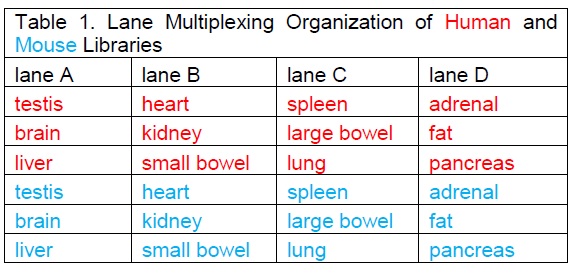
new design
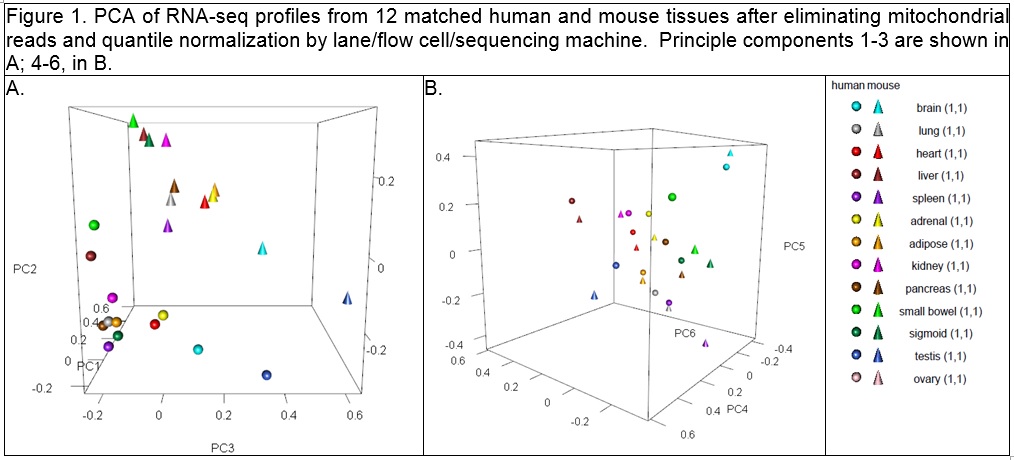
new pca
The new sequencing design seems much more reasonable, and the PCA plot has many characteristics of the original one from the original comparative analysis by Mouse ENCODE (see above), in that yes, there are species specific differences, but there also appears to be a way to collapse along PC2 and PC3 where the tissues will line up with each other, which I kind of would expect.